
A Detailed Look At The 5 Factors Of Low Voltage
The following blog post is taken directly from a Senergy Facebook Livestream with Dr. Jerry Tennant. Watch the video below! The 5 Factors of Low



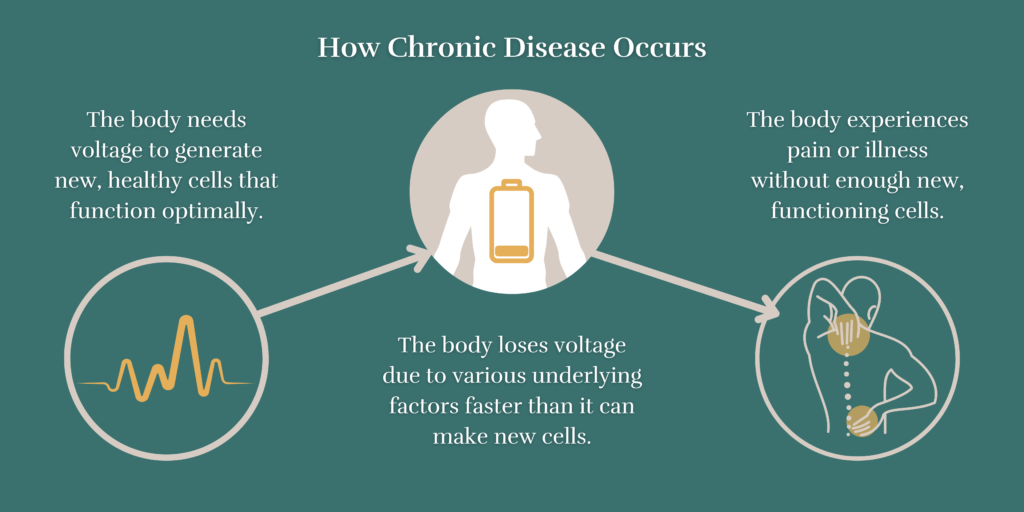













The following blog post is taken directly from a Senergy Facebook Livestream with Dr. Jerry Tennant. Watch the video below! The 5 Factors of Low

What Supplements Are Good For Lupus? If you or someone you know has lupus, it’s not surprising that the patient would be on a quest

How To Strengthen Your Immune System Taken from a Senergy Friday Livestream Interview with Tennant Products director and product developer Scott Jessen. To watch the